| Creating your spreadsheet |
The Tab2MAGE spreadsheet structure is split into three main sections: Experiment, Protocol and Hybridization. The sections are separated from each other by one or more blank lines. Please note that blank lines are used to demarcate the ends of sections, and as such they should not be used within the body of any section. We will discuss each of these three sections in turn.
A series of real-world and conceptual examples is given at the end of this page.
This section holds all of the top-level information about an experiment. It consists of two columns; the left column contains a series of predefined row tags, while the right column contains the actual values pertaining to your experiment. An example is shown below:
| Experiment section | |
| domain | ebi.ac.uk |
| accession | E-EXML-1 |
| quality_control | dye_swap_quality_control |
| experiment_design_type | strain_or_line_design |
| name | <your experiment title> |
| description | <short description of your experiment> |
| release_date | 2004-08-30 |
| submission_date | 2004-07-28 |
| submitter | John Falstaff |
| organization | Windsor Laboratories |
| publication_title | <your manuscript title> |
| authors | John Falstaff; Robin Goodfellow |
| journal | Nature Genetics |
| volume | 12 |
| issue | 4 |
| pages | 123-456 |
| year | 2004 |
| pubmed_id | 12345678 |
With the exception of the section header, all the tags above are optional. However, if the domain or accession tags are not present suitable placeholders will be used instead. A more detailed explanation of the meaning of each of these tags may be obtained by clicking on them.
[ Back ][ Top of page ]
The Protocol section allows the user to define the protocols used in the experiment. The section consists of three required columns (accession, text and name) with an optional parameters column. A fifth column, "type" is available for adding custom ProtocolType terms from the MGED Ontology to your protocols, but its use is not recommended for the majority of cases. This column should generally be omitted, and the tab2mage script will use a set of default protocol types based on how the protocols are used in the Hybridization section.
| Protocol section | |||
| accession | text | name | parameters |
| P-EXML-1 | Cells were grown in YPD (1% yeast extract/2% peptone/2% glucose) to an OD600 of approximately 0.8 | Yeast growth | growth temperature (degree_C); pH |
| P-EXML-2 | <protocol text> | Yeast cell harvesting | pellet weight (mg) |
| P-EXML-3 | <protocol text> | Cell lysis and RNA prep | |
| P-EXML-4 | <protocol text> | cDNA labeling | |
| P-EXML-5 | <protocol text> | Hybridization | hyb temp (degree_C); hyb volume (uL) |
| P-EXML-6 | <protocol text> | Scanning | |
| P-EXML-7 | <protocol text> | Image analysis | |
| P-EXML-8 | <protocol text> | Normalization |
Note that you may omit protocols which have been previously submitted to ArrayExpress, and simply refer to those protocols directly in the Hybridization section. If no new protocols are needed then the entire Protocol section may be omitted. A more detailed explanation of the meaning of each of the fields in this section may be obtained by clicking on the column headings.
[ Back ][ Top of page ]
The Hybridization section describes how each sample links to each hybridization, scanning and subsequent normalization, and as such is the largest and most complex section. There is a simple principle underlying this section, however: Everything which appears on a given line must be related in some way.
The column headings available in this section can be divided into several different types for the purpose of clarity. Note that the various columns in this section may appear in any order, and so you may find that re-ordering the columns makes the spreadsheet more legible for your own application.
For each of the following tables, a more detailed explanation of the meaning of each of the column headings may be obtained by clicking on them.
Names of
materials and processes
These columns are provided so
that you can give unique names to each of the materials used in
your experiment (e.g., samples, extracts, labeled
extracts). Hybridization and normalization events are also given
names. The use of these names is entirely optional, as Tab2MAGE
will attempt to link all of these things together for you. If,
however, your experiment involves complex pooling or splitting
operations then it is recommended that you make use of the
names. Doing so will allow you a much greater degree of control
over the output MAGE-ML.
The following table is an example of a simple reference design in which individual samples are hybridized against a common reference pool:
| Hybridization section | ||||||
| BioSource | Sample | Extract | LabeledExtract | Dye | Hybridization | Normalization |
| S288C | S288C sample | S288C extract | S288C LE | Cy3 | S288C Hyb | S288C Norm |
| S288C | S288C sample | Reference extract | Reference LE | Cy5 | S288C Hyb | S288C Norm |
| Sigma1278b | Sigma1278b sample | Sigma1278b extract | Sigma1278b LE | Cy3 | Sigma1278b Hyb | Sigma1278b Norm |
| Sigma1278b | Sigma1278b sample | Reference extract | Reference LE | Cy5 | Sigma1278b Hyb | Sigma1278b Norm |
| W303a | W303a sample | W303a extract | W303a LE | Cy3 | W303a Hyb | W303a Norm |
| W303a | W303a sample | Reference extract | Reference LE | Cy5 | W303a Hyb | W303a Norm |
Note that Tab2MAGE does not constrain you to use the standard experimental layout:
biosource ----------> sample ----------> extract ----------> labeled extract ----------> hybridization event
[growth, treatment] [extraction] [labeling] [hybridization]
If, for instance, your experiment starts with a series of extracts provided by an external supplier, the sample and extract stages may be omitted:
biosource ----------> labeled extract ----------> hybridization event
[labeling] [hybridization]
Material
types
These columns simply indicate the type of each
material used. The terms should all be instances of MaterialType
from the MGED
ontology. Typical terms are shown in this example:
| Hybridization section | |||
| BioSourceMaterial | SampleMaterial | ExtractMaterial | LabeledExtractMaterial |
| whole_organism | whole_organism | total_RNA | synthetic_DNA |
| whole_organism | organism_part | total_RNA | synthetic_RNA |
| organism_part | cell | polyA_RNA | synthetic_RNA |
These columns are not absolutely required for Tab2MAGE to run. However, the strictures of the MAGE model mean that if these values are not given, then suitable default values must be generated. It is therefore wise to use these columns wherever possible.
Material
characteristics
Each biosource used in the experiment
can have an arbitrary number of characteristics. The
"BioMaterialCharacteristics[]" heading provides a way to
define as many of these characteristics as are needed. The general form of this heading is:
BioMaterialCharacteristics[<category>]
where <category> is a subclass of the BioMaterialCharacteristics class within the MGED ontology. Examples are given in the table below:
| Hybridization section | ||
| BioMaterialCharacteristics[Genotype] | BioMaterialCharacteristics[Organism] | BioMaterialCharacteristics[StrainOrLine] |
| CAD1::myc9:TRP1 | Saccharomyces cerevisiae | S288C |
| CAD1::myc9:TRP1 | Saccharomyces cerevisiae | W303a |
| RTG3::myc18:TRP1 | Saccharomyces cerevisiae | S288C |
| RTG3::myc18:TRP1 | Saccharomyces cerevisiae | W303a |
If the BioSource name column is not used, then the set of characteristics of each biosource is used to determine how many biosources should be created. This means that if all your biosources share identical characteristics, and none are given names, then they will be treated as a single biosource in the output MAGE-ML. To circumvent this behaviour, please name your biosources.
Links to
defined protocols and parameters
This set of columns
allows you to link the protocols defined in the previous section to your hybridizations:
| Hybridization section | |||||
| Protocol[grow] | Protocol[treatment] | Protocol[extraction] | Protocol[labeling] | Protocol[hybridization] | Protocol[scanning] |
| P-EXML-1 | P-EXML-2 | P-EXML-3 | P-EXML-4 | P-EXML-5 | P-EXML-6 |
| P-EXML-1 | P-EXML-2 | P-EXML-3 | P-EXML-4 | P-EXML-5 | P-EXML-6 |
| P-EXML-1 | P-EXML-2 | P-EXML-3 | P-EXML-4 | P-EXML-5 | P-EXML-6 |
| P-EXML-1 | P-EXML-2 | P-EXML-3 | P-EXML-4 | P-EXML-5 | P-EXML-6 |
If you have assigned parameters to your protocols, you may set the parameter values here:
| Hybridization section | ||||
| Parameter[growth temperature] | Parameter[pH] | Parameter[pellet weight] | Parameter[hyb temp] | Parameter[hyb volume] |
| 30 | 6 | 10 | 56 | 50 |
| 31 | 6 | 12 | 56 | 50 |
| 30 | 6 | 10 | 55 | 50 |
| 31 | 6 | 12 | 55 | 50 |
The general scheme linking protocols with experimental stage is as shown below:
biosource ----------> sample --------> extract -------> labeled extract -------> hybridization
[grow, treatment] [extraction] [labeling] [hybridization] |
|
|
----------------------------------------------------------------------------------
|
|
v
hybridization ------------> image -----------> raw data file -----------> normalized data file
[scanning] [image_analysis] [normalization]
Note that for protocols linked to the biological materials, the existence of the protocol implies the existence of the target material. For example, if a growth or treatment protocol is used in a given row then a sample object will be automatically created for that row in the output MAGE-ML.
Experimental
factors
Each of your hybridizations should have one or
more experimental factor values associated with it. These
factors refer to the different conditions used in the
preparation of each hybridization. They may reflect variation in
the starting biosources (BioMaterialCharacteristics) or differences in
the way those biosources have been treated. The general form of this heading is:
FactorValue[<category>]
where <category> is a subclass of the ExperimentalFactorCategory class within the MGED ontology (Note: you are likely to need to descend several subclasses into the heirarchy to find a biologically meaningful category). Examples are given in the table below:
| Hybridization section | ||
| FactorValue[Organism] | FactorValue[Sex] | FactorValue[OrganismPart] |
| Homo sapiens | male | liver |
| Homo sapiens | male | kidney |
| Homo sapiens | female | liver |
| Homo sapiens | female | kidney |
| Mus musculus | male | liver |
| Mus musculus | male | kidney |
| Mus musculus | female | liver |
| Mus musculus | female | kidney |
Your spreadsheet may contain as many "FactorValue[]" columns as necessary to fully describe your experiment.
Data files and
array information
The final class of headings is used to
specify which data files relate to which hybridization. There
are also columns in this section for linking the data files to
the array design used. Here is an example of these columns for a
spotted array:
| Hybridization section | |||
| File[raw] | File[normalized] | Array[accession] | Array[serial] |
| Data1.txt | NormData1.txt | A-EXML-1 | 244532 |
| Data2.txt | NormData2.txt | A-EXML-1 | 244533 |
| Data3.txt | NormData3.txt | A-EXML-1 | 244534 |
| Data4.txt | NormData4.txt | A-EXML-1 | 244535 |
Below is an example of what is required for an Affymetrix submission. Note that for spreadsheet submissions to ArrayExpress we do not require you to upload the actual CDF library file, unless you are using a custom array. For such submissions the "Array[serial]" column should contain Affymetrix Chip Lot numbers:
| Hybridization section | |||||
| File[raw] | File[normalized] | File[exp] | File[cdf] | Array[accession] | Array[serial] |
| Data1.CEL | Data1.CHP | Data1.EXP | YG_S98.cdf | A-AFFY-27 | 2125576 |
| Data2.CEL | Data2.CHP | Data2.EXP | YG_S98.cdf | A-AFFY-27 | 2103523 |
| Data3.CEL | Data3.CHP | Data3.EXP | YG_S98.cdf | A-AFFY-27 | 2468372 |
| Data4.CEL | Data4.CHP | Data4.EXP | YG_S98.cdf | A-AFFY-27 | 2542678 |
Currently supported data file formats include GenePix, Affymetrix, Agilent, ScanArray/QuantArray, ScanAlyze and Arrayvision. The MetaColumn/MetaRow format favoured by ArrayExpress is of course also supported.
The "Array[accession]" column is required in any spreadsheets referencing data files. If Tab2MAGE is used in the absence of data files then the array information may be omitted, although such usage of Tab2MAGE will obviously not be acceptable as an ArrayExpress data submission.
If you prefer to process your normalized data in a single file mapping to multiple hybridizations, Tab2MAGE also supports including this data as a Final Data Matrix file. This file can be included in an experiment using the "File[transformed]" column. Please note that the final data matrix file must be formatted as described in the data file notes.
| Hybridization section | |||
| File[raw] | File[transformed] | Array[accession] | Hybridization |
| Data1.txt | FinalDataMatrix.txt | A-EXML-1 | Hyb1 |
| Data2.txt | FinalDataMatrix.txt | A-EXML-1 | Hyb2 |
| Data3.txt | FinalDataMatrix.txt | A-EXML-1 | Hyb3 |
| Data4.txt | FinalDataMatrix.txt | A-EXML-1 | Hyb4 |
Please see the MAGE mappings for other supported column headings, and further information on how the spreadsheet information is used to create the MAGE-ML document.
[ Back ][ Top of page ]
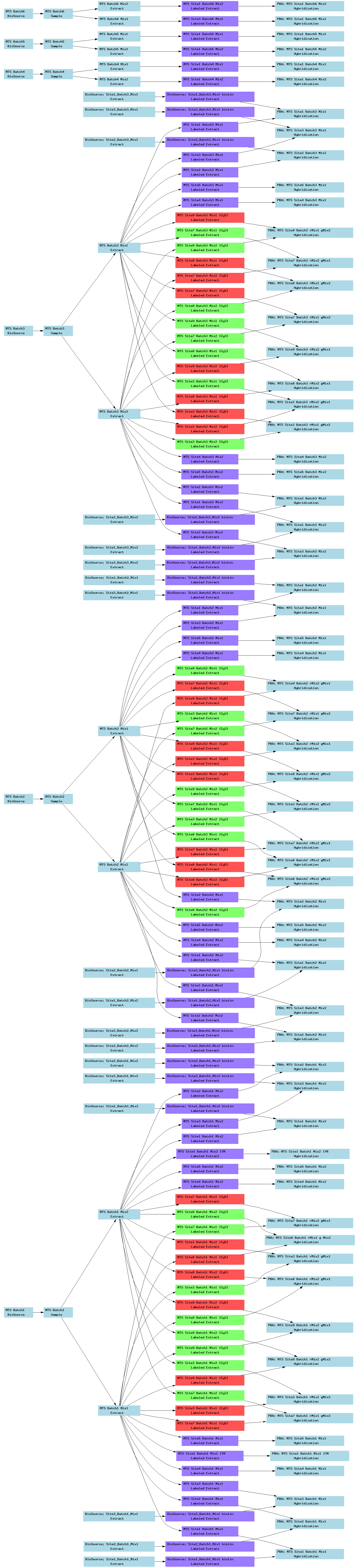
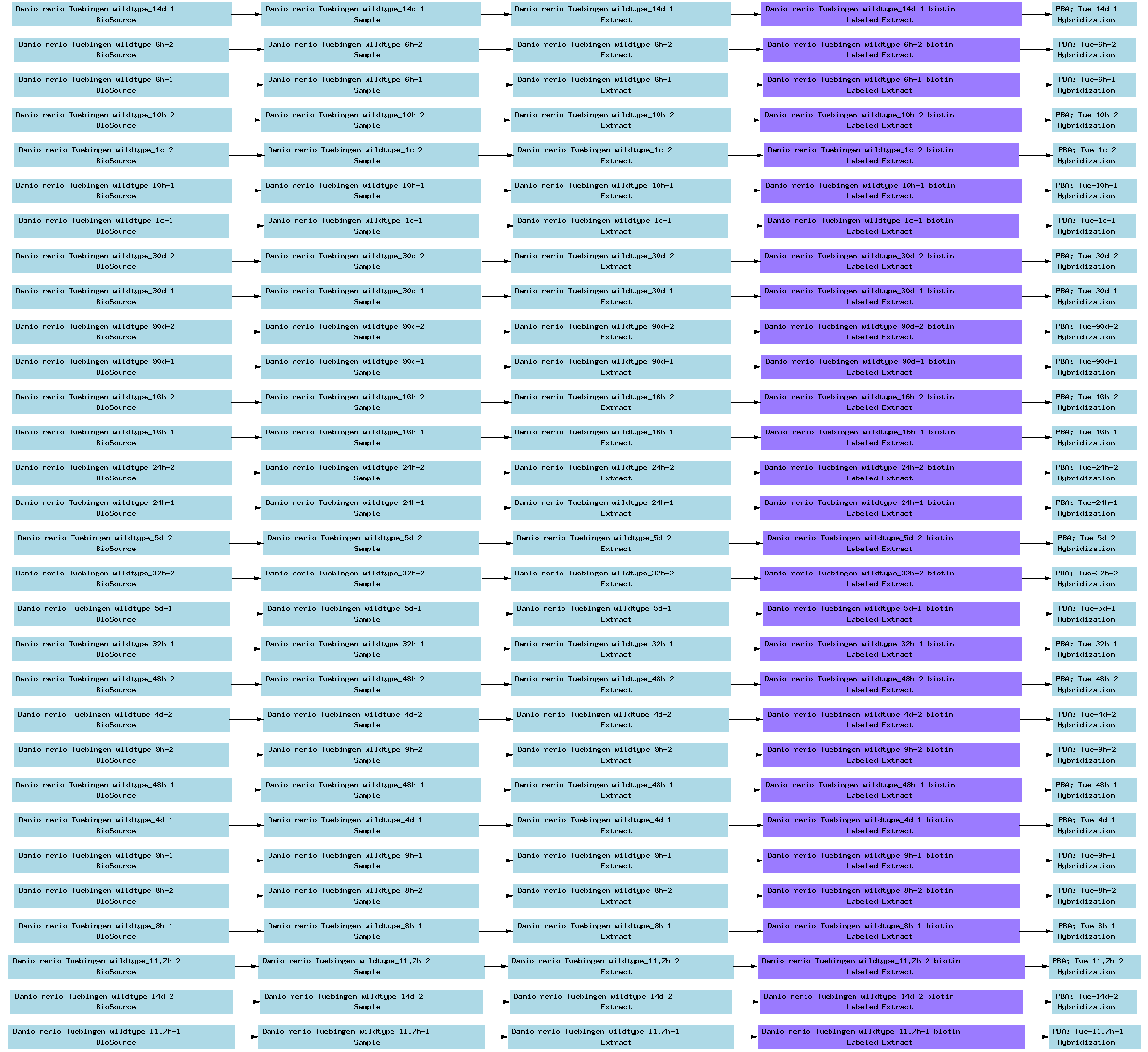
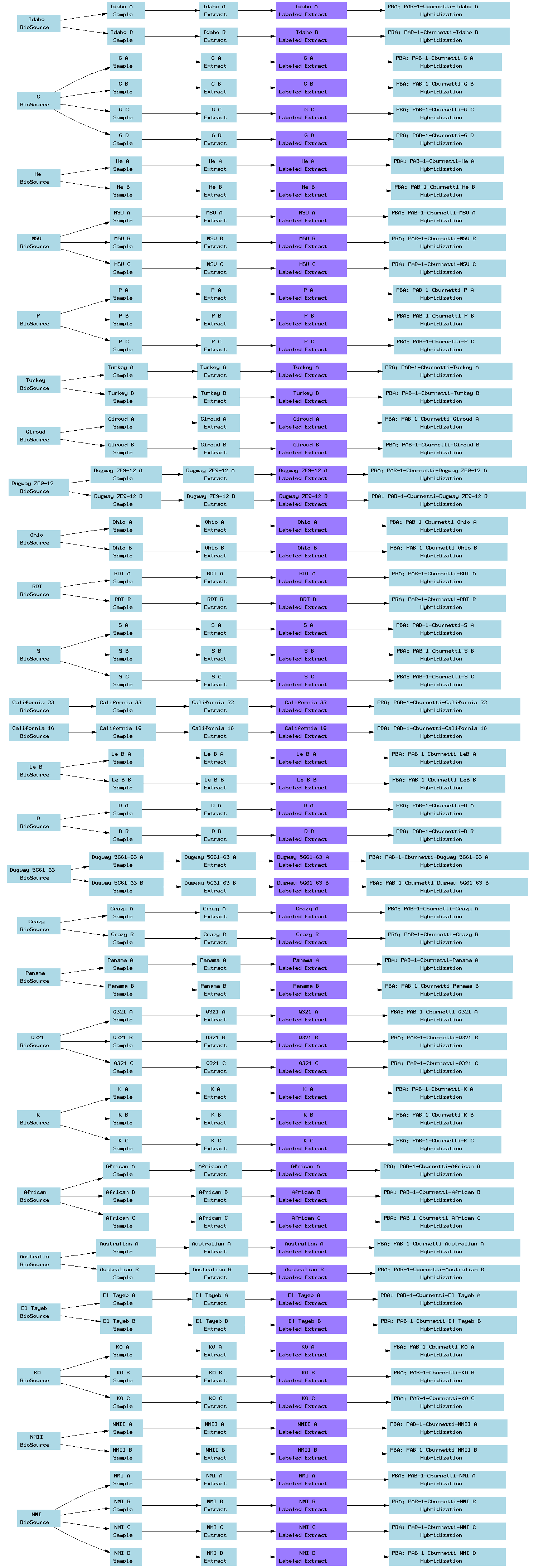
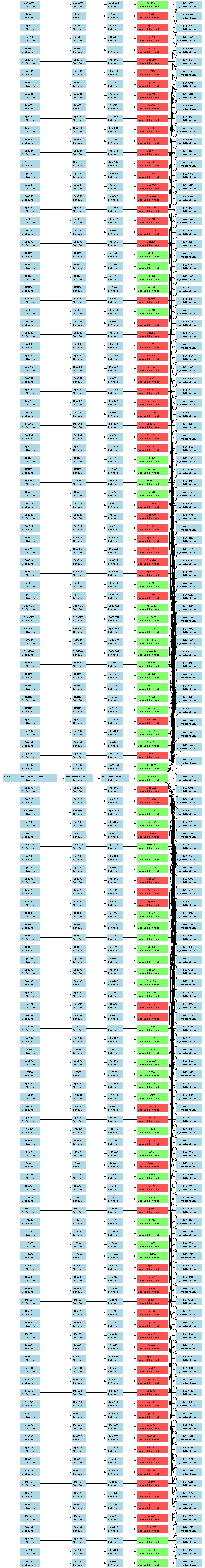
Below is a selection of some of the real-world curated Tab2MAGE spreadsheets submitted to ArrayExpress over the past few months:
| Spreadsheet | Graph | Experiment name | Platform | Link to experiment |
|---|---|---|---|---|
| E-TABM-16.txt | PNG | FDA-CDER MTS RNA reagent cross platform test | Multiple platforms | ArrayExpress |
| E-TABM-18.txt | PNG | Transcription profiling of 35 different Arabidopsis thaliana ecotypes | Affymetrix | ArrayExpress |
| E-TABM-22.txt | PNG | Transcription profiling of human lung cancers and lung cancer cell lines miRNA expression | One-channel custom array | ArrayExpress |
| E-TABM-33.txt | PNG | Transcription profiling of zebrafish development | Affymetrix | ArrayExpress |
| E-TABM-35.txt | PNG | Comparative genomic hybridization of 25 Coxiella burnetii isolates relative to the Nine Mile (RSA493) reference isolate | Affymetrix | ArrayExpress |
| E-TABM-54.txt | PNG | Comparative genome hybridization of 137 Bordetella pertussis strains | Two-color custom array | ArrayExpress |
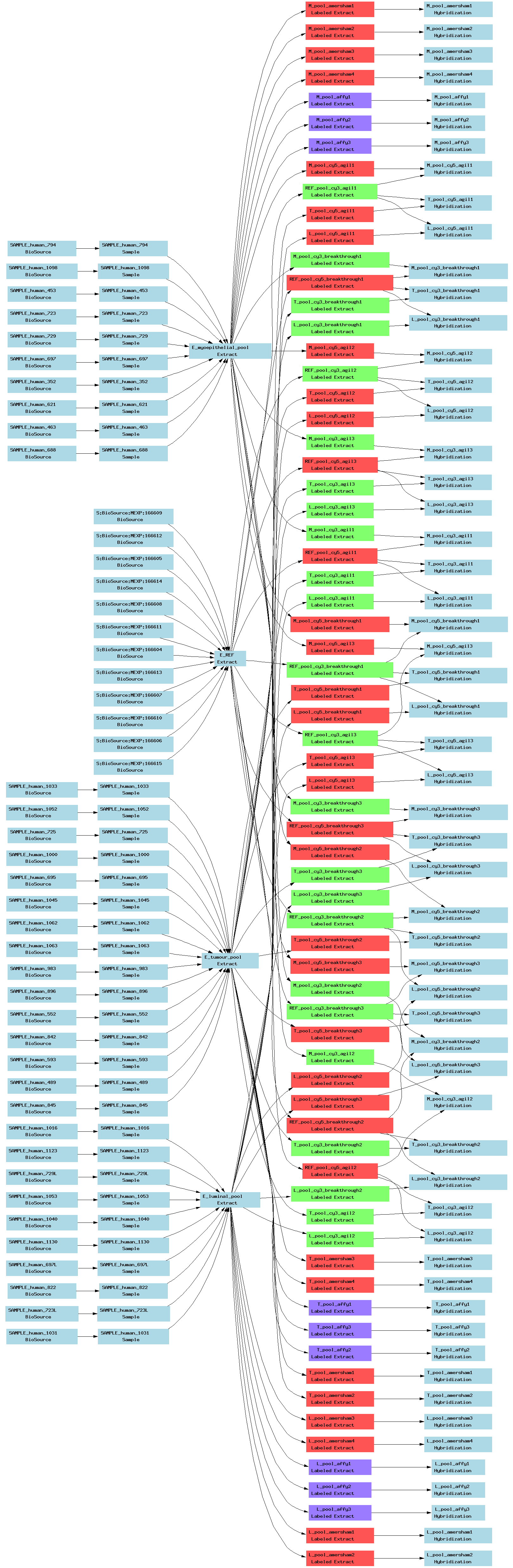
| E-TABM-66.txt | PNG | Transcription profiling of normal and malignant human breast epithelial cells | Multiple platforms | ArrayExpress |
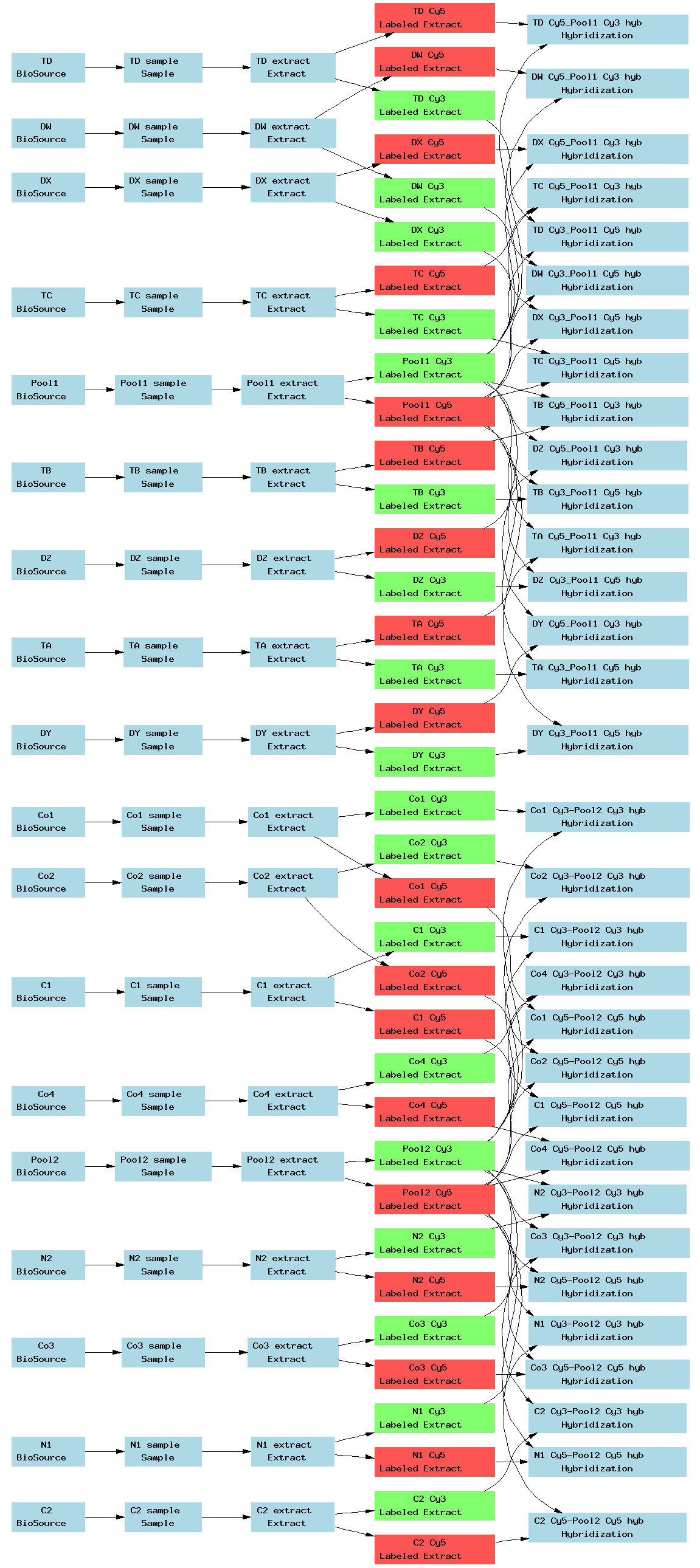
| E-TABM-70.txt | PNG | Transcription profiling of human cell lines treated with cytochalasin D and nocodazole with the aim of characterising tetraploid clones | Agilent | ArrayExpress |
| E-TABM-102.txt | PNG | Transcription profiling of wild type and ATF3 -/-mouse bone marrow macrophages stimulated with lipopolysaccharide over time | Affymetrix | ArrayExpress |
| E-TABM-134.txt | PNG | WGA-LCM and Genomewide Survey of Lung Cancer | Affymetrix | ArrayExpress |
| E-TABM-136.txt | PNG | Transcription profiling of human and chimpanzee heart, brain, testis and lymphblastoid cell lines to study functionality of intergenic transcription | Affymetrix | ArrayExpress |
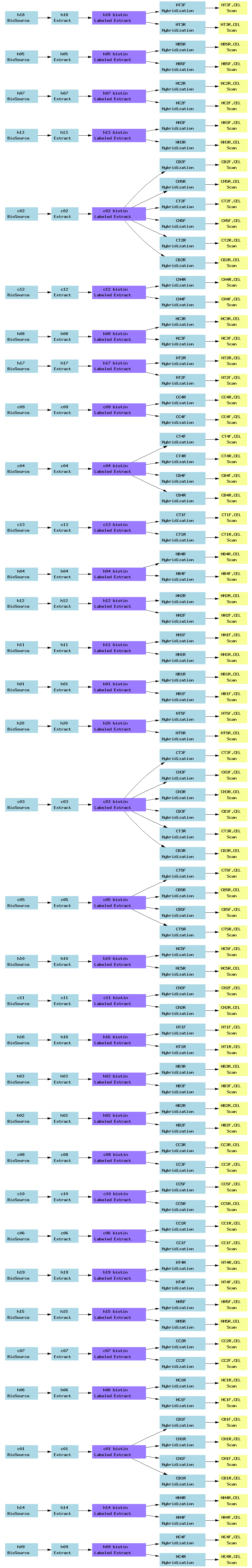
| E-TABM-140.txt | PNG | Chromatin immunoprecipitation (ChIP-chip) of human erythroleukemia cell line K-562 with anti-histone antibodies using an ENCODE array | Two-color custom array | ArrayExpress |
| E-TABM-163.txt | PNG | Transcription profiling of murine presomitic mesoderms of 17 samples at various time points to identify cyclic genes of the mouse segmentation clock | Affymetrix | ArrayExpress |
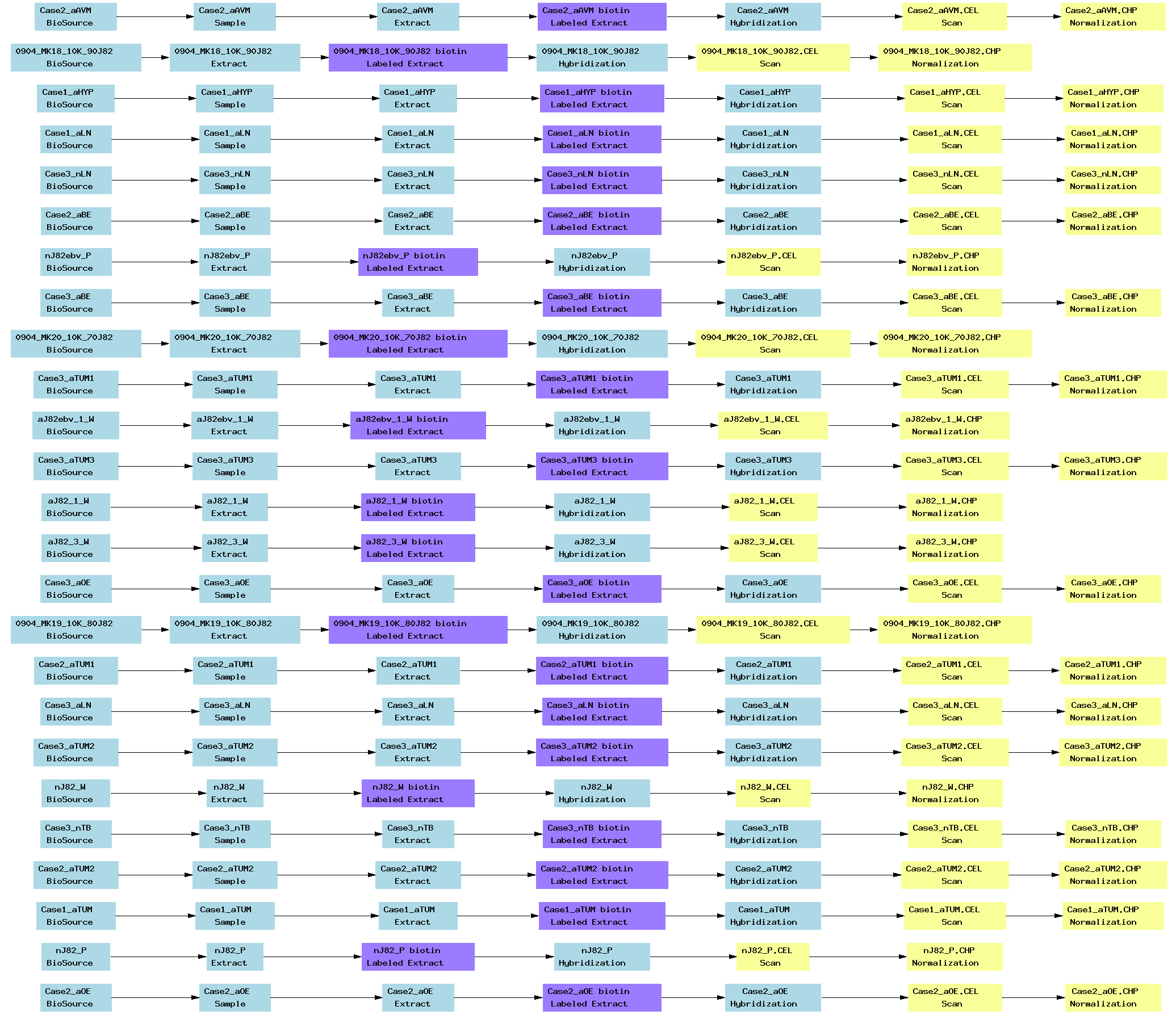
| E-MEXP-880.txt | PNG | Methylation profiling of normal and cancerous breast cells from human patients and cell lines in a 125 kB region of the HOXA cluster | Two-color custom array | ArrayExpress |
[ Back ][ Top of page ]
A series of very simple conceptual examples is given here:
[ Back ][ Top of page ]
Please see the experiment checker and tab2mage.pl help notes for more information.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}